
- Home
- Companies
- Sibylla Biotech S.p.A.
- Products
- Sibylla - Computational Platform
Sibylla - Computational Platform
What makes us unique is the power of our computational platform, which enables us to reconstruct the series of conformational rearrangements underlying the process of protein folding at an atomistic level of detail.
In contrast, our algorithms are capable of driving the polypeptide chain through the conformational landscape, overcoming the process of trial and error responsible for the timescale-inaccessibility typical of protein folding. Our scheme enables us to accurately characterize the folding pathways of proteins with size up to 400 amino acids in less than a month.
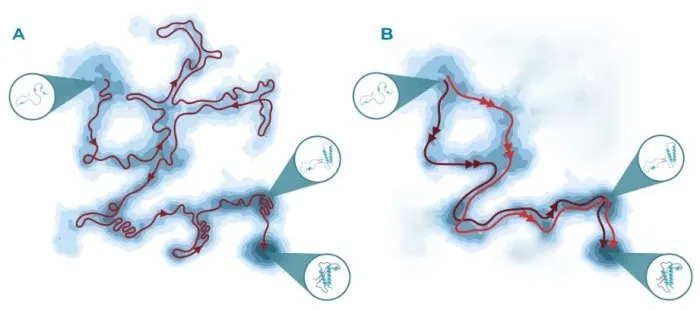
Our sole requirement to fully characterize the folding mechanism of a target protein is the structure of its native state. This is used by our approach to generate a collective variable that defines the reaction progress. Our algorithm relies on such information to drive the dynamics by employing a custom implementation of adiabatic-biased molecular dynamics.
We then ensure the reliability of the sampled pathways by using filtering schemes derived from theoretical physics. Density-based clustering algorithms are then employed to deliver a network of states and transitions accurately representing the mechanism, from which relevant folding intermediates can be identified. Once the candidate structures are selected, we scout their surfaces to identify druggable pockets which are absent in the native state. These sites can be used as the starting point of an in-house virtual screening campaign, eventually delivering promising hit compounds for the target of interest.
Our platform is powered by a large GPU cluster, able to run molecular dynamics simulations with a computing performance of hundreds of TFLOPS.

We are continuously improving our algorithms and developing new techniques to push innovation forward. The reconstruction of the protein folding mechanism can be improved by taking into account the complexity of the cellular environment in which proteins acquire their native fold. One of the key elements of folding in vivo is that it occurs sequentially during protein synthesis. Indeed, vectoriality can have a significant influence on the folding mechanism of proteins and should be taken into consideration for a more accurate characterization of folding intermediates. Here at Sibylla we are developing dedicated algorithms that for the first time are capable of simulating the folding process during synthesis by the ribosome and translocation across the endoplasmic reticulum membrane. This will allow us to improve the accuracy and reliability of our predictions.
At the same time, we are investing our research efforts in expanding the range of druggable targets developing algorithms for the identification of so-called “cryptic pockets”. These sites represent potentially valid ligandable candidates for therapeutic purposes, but have the characteristics of not being immediately apparent in unbound protein native states. We are able to detect the presence of these sites by means of enhanced conformational exploration techniques.
